Research summary
We concentrate on several areas of molecular simulation:
Design of wide-coverage polarizable Force Fields.
Development of MD tools that properly contain our advanced Force Fields.
Implementation and development of advanced sampling methods for determination of protein-ligand binding affinities.
Modelling the liquid state with the emphasis on aqueous biological environments as well as ionic liquids and electrolytes.
Selected Publications
Our publications announce major milestones in the creation and refinement of our simulation tools
Combining Force Fields and Neural Networks for an Accurate Representation of Chemically Diverse Molecular Interactions Journal of the American Chemical Society (2023). This is our flagship publication. It introduces AI into some parts of the model. We have reached our goal: the models can accurately describe and predict properties of biological, ionic and other complex systems.
The determination of free energy of hydration of water ions from first principles (2024). In Press. To show and test the power of our methods we describe the most difficult ion of all - the water ion(s). As a test, without any approximations we predict the pH of water correctly.
Combining Force Fields and Neural Networks for an Accurate Representation of Bonded Interactions The Journal of Physical Chemistry A (2024). Our NN fingerprint is applied to bonded interactions.
Neural Network corrections capture the energetics of Nuclear Quantum Effects in the condensed phase. Journal of Chemical Theory and Computation (2023). This paper applies our NN formalism to capture and describe the quantum nature of nuclear motion.
Protein–Ligand Binding Free-Energy Calculations with ARROW─A Purely First-Principles Parameterized Polarizable Force Field Journal of Chemical Theory and Computation (2022). This paper describes a novel sampling method, and establishes that the Arrow2 ForceField is insufficient to describe certain strong interactions.
Accurate determination of solvation free energies of neutral organic compounds from first principles. Nature Communications 13.1 (2022): 1-7. This paper demonstrates the accurate prediction of liquid state properties by using models parameterized by QM calculations only.
On the importance of accounting for nuclear quantum effects in ab-initio calibrated force fields in biological simulation. Proceedings of National Academy of Sciences, 100 (1), 119-124 (2018). This paper emphasizes that accurate simulations must account for non-classical nature of nuclear motion.
Prediction of cyclohexane-water distribution coefficient for SAMPL5 drug-like compounds with the QMPFF3 and ARROW polarizable force fields Journal of Computer-Aided Molecular Design, 30 (11), 977–988 (2016). Our first foray into blind prediction of solvation (beta tools and models).
Polarizable Force Fields for Proteins. In: Náray-Szabó G. (eds) Protein Modelling. Springer, Cham. 91-134 (2014). A review of polarizable force fields.
Molecular Recognition in Pharmacology Mikhail Darkhovsky. CRC Press, Routelage, Taylor and Francis ( 2024). A book on Pharmacology written by one of the members of our team. Congratulations Mikhail!

Protein-ligand binding
LSD in a protein receptor
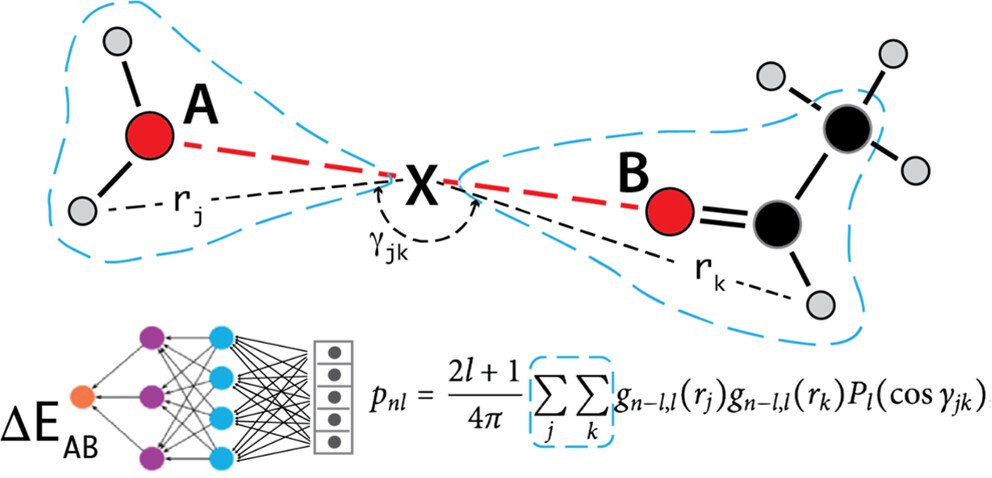
A novel Neural Network encoding for accurate molecular interactions
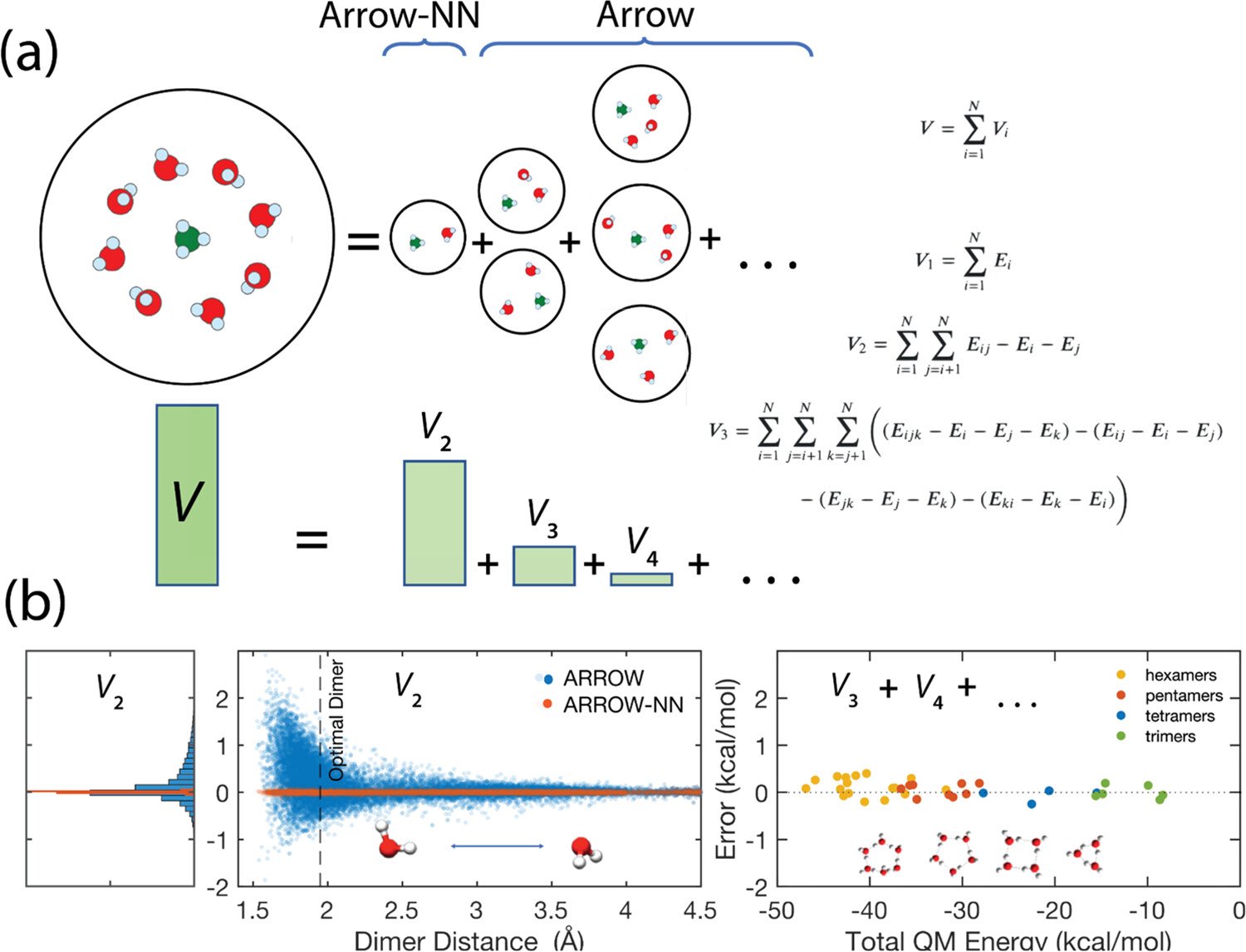
Using physics to greatly enhance inference and robustness of models

Our models are the first to be truly predictive

Inference to an arbitrary number of molecules is done with Physics.

Arbitrary liquid mixtures correctly modeled.

Pharmacology textbook by Mikhail Darkhovskiy